Designing drugs that can help 30 million adults and children living with HIV is an ongoing battle made worse by the virus’s ability to develop resistance to some existing drugs.
Researchers are using x-ray crystallography at the Australian Synchrotron to help identify potential new drugs to address the resistance problem. For example, Jerome Wielens from St Vincent’s Institute in Melbourne recently used the Australian Synchrotron to obtain valuable new clues about different kinds of chemical compounds that could potentially be more effective against HIV.
Infection with human immunodeficiency virus (HIV) leads to acquired immunodeficiency syndrome (AIDS). This debilitating disease attacks and eventually sabotages the immune system, ultimately resulting in death. Treatments are available for HIV that reduce viral loads and slow the progression of AIDS but none of these eradicate the virus or prevent the spread of infection. Moreover, these treatment regimes are being compromised by the emergence of drug-resistant strains of HIV.
Although the development of effective treatments has helped reduce the death rate, AIDS-related causes still account for about 1.8 million deaths a year. HIV/AIDS is the second most common cause of death for people aged 20 to 24, according to UK-based international HIV and AIDS charity AVERT.
Drugs that block the action of an HIV protein called integrase are an important part of current treatments for HIV/AIDS. HIV-1 integrase is an essential protein for HIV replication and the development of AIDS. It catalyses the insertion of the viral DNA genome into the host DNA, which establishes the HIV infection in the host’s body. Preventing integrase from performing this function stops the virus replicating and halts the progression to AIDS.
The first integrase inhibitor approved for clinical use is raltegravir. Several other inhibitors in advanced clinical development act via the same mechanism (i.e. they bind to the same site on integrase) as raltegravir. However, strains of HIV that are resistant to raltegravir are also resistant to some of these preclinical drug candidates. As a result, there is significant interest in identifying other potential drug-binding sites on integrase.
Structure-based drug design

Jerome Wielens (St Vincent’s Institute) prepares to use the MX2 beamline at the Australian Synchrotron.
One of life's essential building blocks, proteins perform many tasks including acting as molecular machines, transporters, switches and catalysers, as well as contributing to the overall structure of an organism. X-ray crystallography allows researchers to determine the three-dimensional shape of a protein; they can then use this information to understand how the protein functions – and potentially modulate its activity. Because synchrotron x-rays are much more powerful than laboratory x-rays, they produce far more precise pictures of protein structures.
Structure-based drug design is a method of designing drugs that uses the three-dimensional shape of a protein target to develop compounds that modulate (usually inhibit) the activity of the protein to obtain a desired response. For example, a compound may prevent the protein from performing an essential function for the propagation or survival of a virus, a cancer cell or bacterium, and therefore have the potential to be developed into a treatment for that particular virus, cancer or bacterium.
Fragment-based drug discovery (FBDD) is an approach for identifying which different types of molecules can bind to target proteins and then determining which part of the target protein they actually bind to (usually called a binding site or pocket). ‘Fragments’ are small compounds of relatively low complexity that bind to sub-pockets within a binding site on the protein. X-ray crystallography is used to visualise these fragments bound to the protein of interest so that chemists can develop more-complex compounds that combine different fragments or ‘grow’ them; the aim is that some of these more-complex compounds will bind the protein strongly enough to block its action and ultimately lead to the development of new medicines based on these compounds.
Small steps with big potential
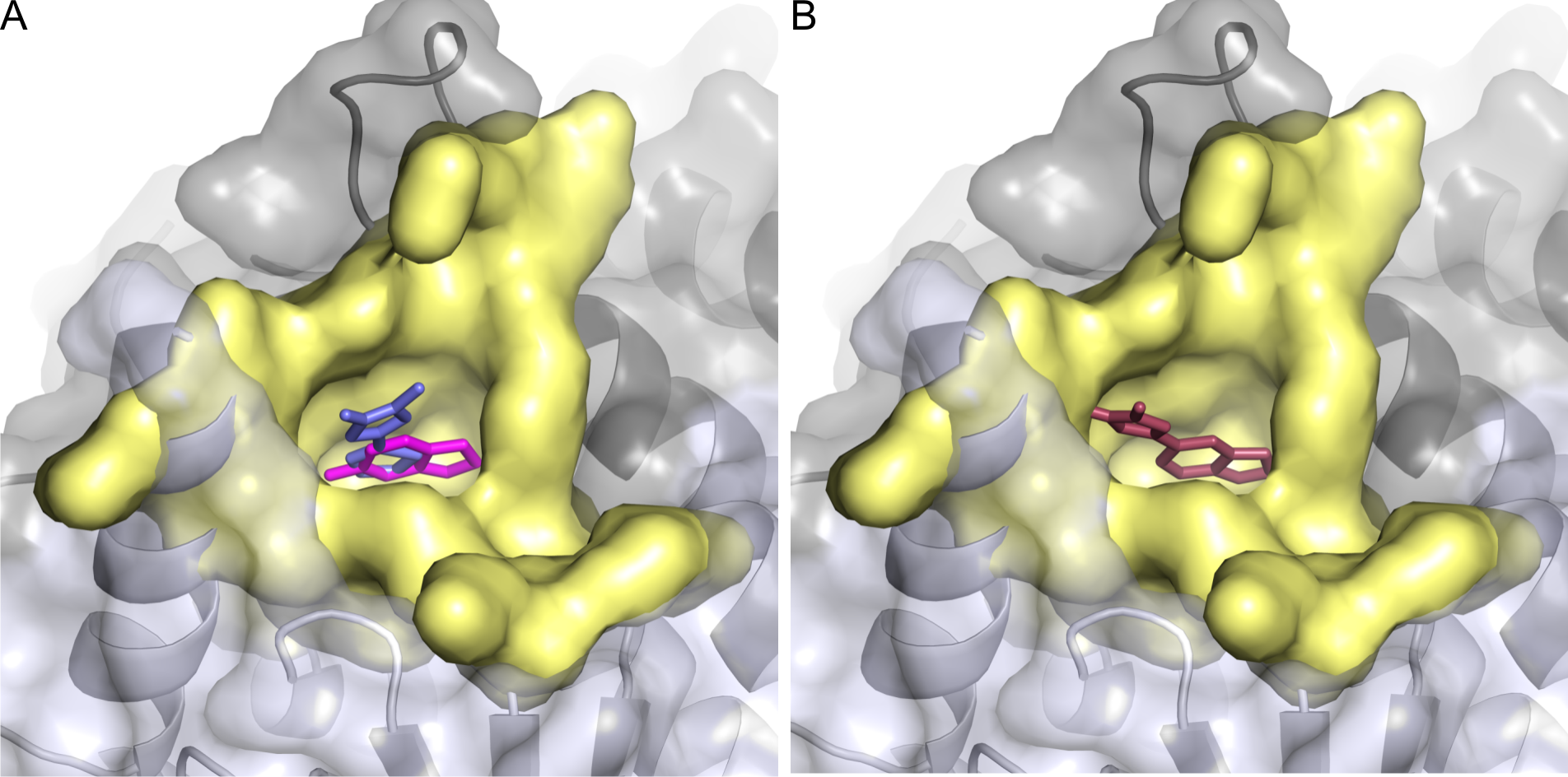
LEFT: Fragment-based drug design involves finding small molecules or ‘fragments’ (two shown here in magenta/blue) that bind to a key protein (in this case HIV-1 integrase from the AIDS virus). RIGHT: Researchers then designed a molecule (shown here in raspberry) that combines the characteristics of these fragments and binds more tightly to the protein. Images: Jerome Wielens. Figures prepared with PyMOL http://www.pymol.org
Jerome Wielens from St Vincent’s Institute in Melbourne and his colleagues and collaborators are working to identify fragment molecules that bind to HIV-1 integrase and determine the three-dimensional structure of these fragments in complex with integrase. They are using a structure-guided approach to design and build more complex molecules with improved binding power.
The researchers initially used nuclear magnetic resonance (NMR) spectroscopy to investigate a library of 500 molecules and identify a series of fragment compounds that bind to integrase. They then used the crystallography beamlines at the Australian Synchrotron to determine the crystal structures of some of these compounds in complex with integrase. The synchrotron information helped the group to elaborate their initial hits and produce compounds with much stronger affinity for integrase (in the micromolar range). In the process, they also identified a previously unknown binding pocket on integrase that has the potential to be a valuable drug target.
The group’s synchrotron results provide a useful basis for designing more potent compounds that bind to integrase and disrupt its functioning.
The work was partly funded by an ARC Linkage grant and done in collaboration with Professor Michael Parker of St Vincent's Institute, Drs David Rhodes and John Deadman of Avexa Ltd and Dr Stephen Headey, Dr David Chalmers and A/Professor Martin Scanlon of Monash Institute of Pharmaceutical Sciences.
Click here to go to a recent research paper authored by Jerome and his colleagues.